
February 28, 2019
https://doi.org/10.23999/j.dtomp.2019.2.2
J Diagn Treat Oral Maxillofac Pathol 2021;3:38–51.
Under a Creative Commons license
HOW TO CITE THIS ARTICLE
Acosta–Guio J, García–Acero M, Jiménez JA, Duque A. Muenke syndrome: variable expressivity between family members. J Diagn Treat Oral Maxillofac Pathol 2019;3(2):38−51.
Contents: Summary | Introduction | Case Report | Discussion | Role of Co-Authors | Fundings | Acknowledgements | References (39)
Summary
Muenke syndrome (MS) is the most common syndromic form of craniosynostosis with an incidence of 1 in 30,000 births, it corresponds to 8 percent of all craniosynostosis.1,2 We report a data of a 5-year-old male patient with syndromic craniosynostosis and his father. Clinical view of patient and his father is analyzed. The 3D computed tomography scans are also discussed. A single surgery has been performed as a treatment for coronal craniosynostosis with bilateral frontoorbital advancement with bone grafts.
A new clinical syndrome is being defined on the basis of the molecular finding in a 61 individuals.
––Maximilian Muenke et al, 19971
USA and UK
Introduction
Craniosynostosis occurs in approximately 1 in 3,000 live births and is characterized by the premature fusion of one or more cranial sutures resulting in malformation of the skull and face. They usually occur as an isolated and sporadic anomaly; however, craniosynostosis is a component in more than 150 described syndromes.
Muenke syndrome (MS) [OMIM #602849], is the most common syndromic form of craniosynostosis with an incidence of 1 in 30,000 births, it corresponds to 8% of all craniosynostosis.1, 2 This condition has autosomal dominant inheritance with incomplete penetrance and variable expressivity, the classic presentation includes unilateral or bilateral craniosynostosis of the coronal suture,3, 4 wide thumbs, carpal and tarsal fusion and presence of the c.749C> G mutation (p.Pro250Arg ) in the FGFR3 gene1, 4-6 affecting more severely women.7 The clinical phenotype is heterogeneous and varies from few detectable clinical manifestations, “isolated” craniosynostosis, to more complex findings that overlap with other classic craniosynostosis syndromes such as Crouzon syndrome, Pfeiffer or Saethre-Chotzen syndrome. Subtle phenotypes with this mutation such as macrocephaly or minor facial anomalies have also been reported.7, 8 Some individuals heterozygous for the mutation in the FGFR3 gene may be clinically and radiographically asymptomatic.9
The spectrum of clinical presentation includes unilateral or bilateral synostosis of the coronal suture, occasionally associated with synostosis of other sutures, resulting in maxillary and orbital hypoplasia, associated to bone involvement of the extremities. In unilateral synostosis, the phenotype is presented with plagiocephaly, facial asymmetry (hypoplasia of the middle facial third, dystopia, palpebral ptosis, ogee palate, cleft lip and/or palate, dental malocclusion and mild retrognathia), flattening of the frontal region and anterior implantation of the ear. In contrast to bilateral synostosis in which the facial phenotype is presented with brachycephaly, bulging of the temporal bone and facial asymmetry.1-3, 5, 6 Conductive hearing loss caused by stenosis or atresia of the external auditory canal and/or malformations of the ossicles has been described6 and sensorineural hearing loss has been related to impaired maintenance of homeostasis of the inner ear.10 Additionally, mild osteopenia, scoliosis and changes in the joints have been described without compromising size.6 Cognitive development is usually normal, although cases with cognitive impairment have been described in relation to the presence of intracranial hypertension, age of surgical correction, socioeconomic status of the family and schooling of the child.1 The oral findings described in patients with MS include dental crowding, malocclusion, crossbite and open bite, probably due to the combined effect of high arched palate and the side effects of craniosynostosis in the skull, facial bones and dental architecture.4
Patients with syndromic craniosynostosis require multidisciplinary management that includes geneticist, ophthalmologist, otorhinolaryngologist, pediatrician, psychologist, maxillofacial surgeon, and speech therapist.4 In severe cases of craniosynostosis with elevated intracranial pressure, the mainly treatment is the surgery. For children with less severe problems, the shaping orthoses can help to reorganize the skull to facilitate the normal growth of the brain and improve the general appearance of the head.11 The surgical correction should be ideally done between 6 to 12 months. The surgical decision should be made based on the aspects that involve: the capacity of brain development for cranial synostosis and the repercussion of intracranial hypertension, as well as the effects generated by mid-facial hypoplasia on the airway, and the commitment of other systems and organs.12
MOLECULAR PHYSIOPATHOLOGY
The family of fibroblastic growth factor receptors (FGFR) plays an important role in the craniofacial development during endochondral ossification of the skull and face, when interrelated with three receptors in the facial mesenchyme. FGFR1 is expressed continuously during the development of the craniofacial complex, FGFR2 is expressed exclusively in the mass of the medial frontonasal mesenchyme, and FGFR3 is expressed in the caudal portion of the frontonasal region and in the lateral portion of the maxillary prominences.3, 4 The molecular defects in FGFR are related to disorders of the bone tissue, due to changes that produce gain-of-function, through a ligand in a dependent or independent way. Mutations in the FGFR1, FGFR2 and FGFR3 genes are mainly associated with syndromic craniosynostosis, in which around 500 different syndromes have been described, among which the syndromes of Apert, Crouzon, Pfeiffer, Beare-Stevenson, Jackson-Weiss, and Muenke are highlighted.2, 3
The mutation c.749C> G (Pro250Arg) responsible for MS is one of the most common transversions in humans,6 with a calculated mutational rate of 8 × 10-6 per haploid genome, in addition, there is evidence of the exclusive paternal origin of the mutation, as well as the association with advanced paternal age.13, 14 Molecular confirmation of the Pro250Arg mutation is not only important for genetic counseling, also for patient management purposes. In the study reported by Thomas et al15 in individuals with unicoronal or bicoronal synostosis and nonspecific phenotype, it was found that those with MS with molecular confirmation tended to require reoperation due to increased intracranial pressure after surgery.
CASE REPORT
We report a 5-year-old male patient, product of first pregnancy of non-consanguineous parents, with history of sensorineural hearing loss in hearing aid management and neurodevelopmental delay. Evidence at physical examination of brachycephaly with temporary overlapping of bones, triangular face, broad forehead, broad metopic suture, facial asymmetry (hypertelorism, oblique palpebral fissures directed downward, scarce eyebrows, palpebral ptosis, mediofacial hypoplasia, depressed nasal bridge), auricular pavilions facing posterior, nasal wing hypoplasia, flat philtrum, high arched palate, microrethrognathia, thin upper lip, (Fig 1A and B). The rest of the physical examination evidenced low height and brachydactyly (Fig 1C).
Computed tomography (CT) with three-dimensional reconstruction of the skull showed bilateral synostosis of the coronal suture (synonym: coronal synostosis), increase of the posterior digital marks, and increase of the space of metopic suture (Fig 2).
Regarding the coexistence of craniosynostosis and brachydactyly, syndromic craniosynostosis was considered. Genomic DNA was isolated from a peripheral blood sample to perform the next-generation sequencing panel of genes associated with syndromic craniosynostosis, this test revealed heterozygous variant c.749C>G (p.Pro252Arg) in the FGFR3 gene, previously reported as pathogenic associated to MS. The study of segregation of the variant, exhibited that the father was the carrier of the variant, physical evaluation revealed (Fig 3) a slight spectrum of the condition with broad forehead, brachycephaly, facial asymmetry (hypertelorism, oblique palpebral fissures directed downward, scarce eyebrows, palpebral ptosis), flat philtrum and thin upper lip.
A single surgery has been performed as a treatment for coronal craniosynostosis with bilateral frontoorbital advancement with bone grafts. Currently, the patient is in multidisciplinary monitoring by geneticist, maxillofacial surgeon, neurosurgeon, endocrinologist, and otorhinolaryngologist. Comprehensive management with language therapies is also indicated.
DISCUSSION
The main characteristic of MS is coronal synostosis that can be unilateral or bilateral. Since the syndrome was described for the first time,1 several cases have been described5, 6, 16 that have contributed to the broader description of the phenotype originally described.
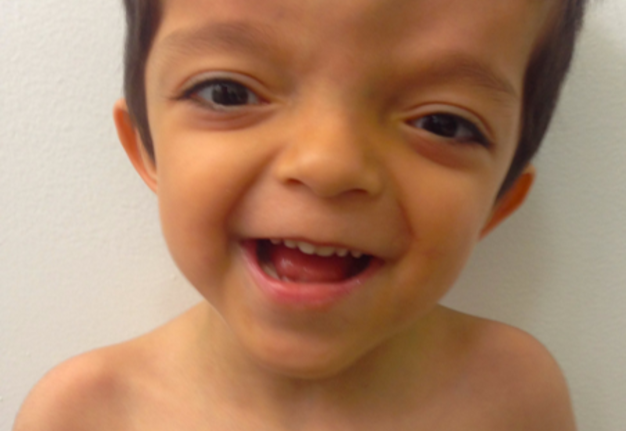
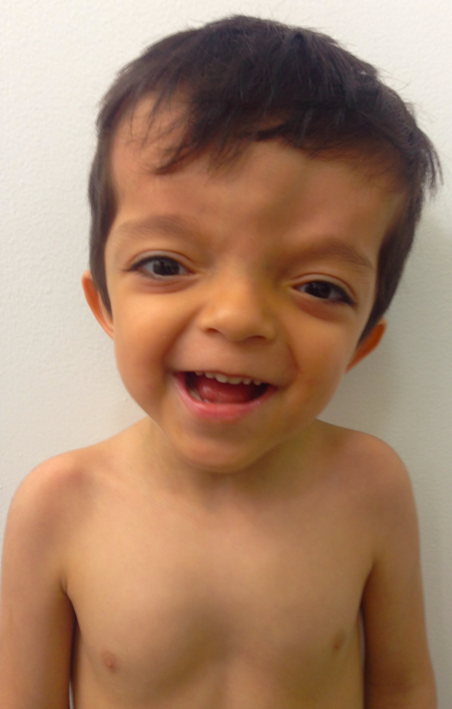
FIGURE 1A. Frontal (A) of a 5-year-old patient with brachycephaly (the shape of a skull shorter than typical skull), triangular face, broad forehead, facial asymmetry (hypertelorism, oblique palpebral fissures directed downward, scarce eyebrows, palpebral ptosis, and depressed nasal bridge), flat philtrum, thin upper lip and micrognathia.

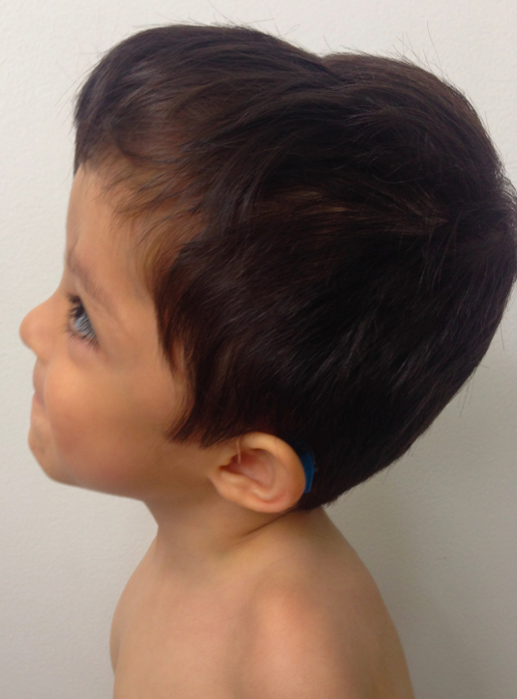
FIGURE 1B. Left side view (B) of a 5-year-old patient with brachycephaly (the shape of a skull shorter than typical skull), triangular face, broad forehead, facial asymmetry (hypertelorism, oblique palpebral fissures directed downward, scarce eyebrows, palpebral ptosis, and depressed nasal bridge), flat philtrum, thin upper lip and micrognathia.

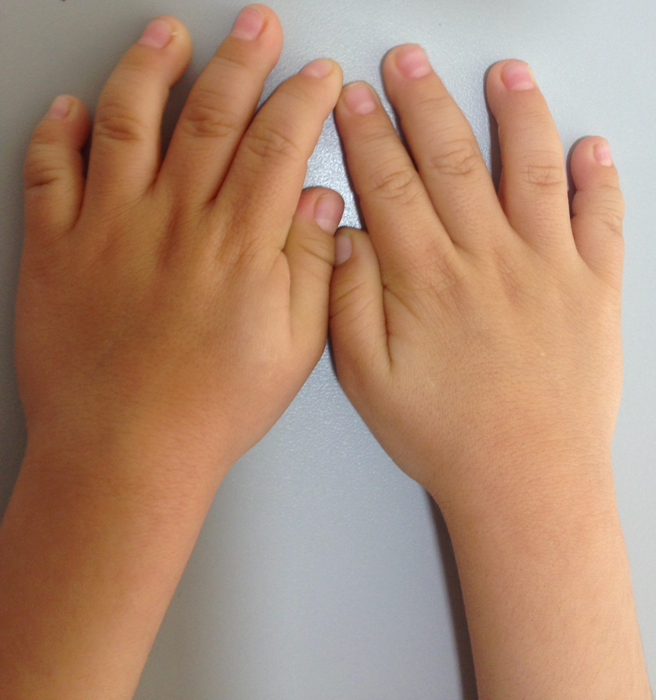
FIGURE 1C. Photograph of a 5-year-old patient’s hands. (C) A brachydactyly (a shortening of the fingers) is noted.
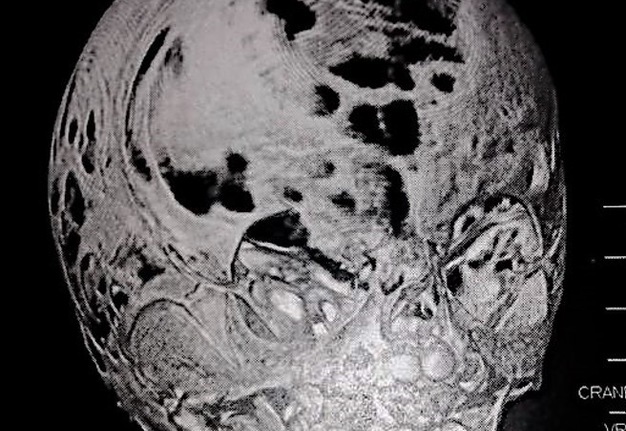
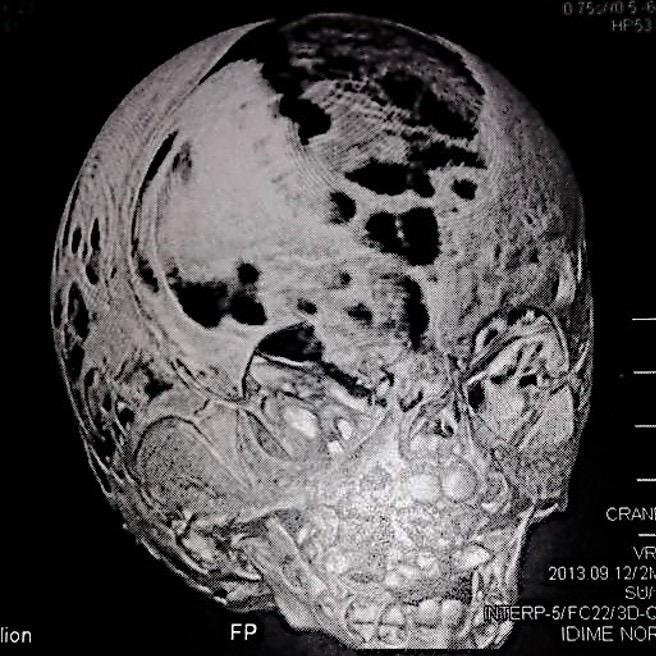
FIGURE 2A. Three-dimensional (3D) reconstruction CT scan of the skull evidence bilateral synostosis of the coronal suture, increase of the posterior digital marks. Noted the increase of the space of metopic suture.
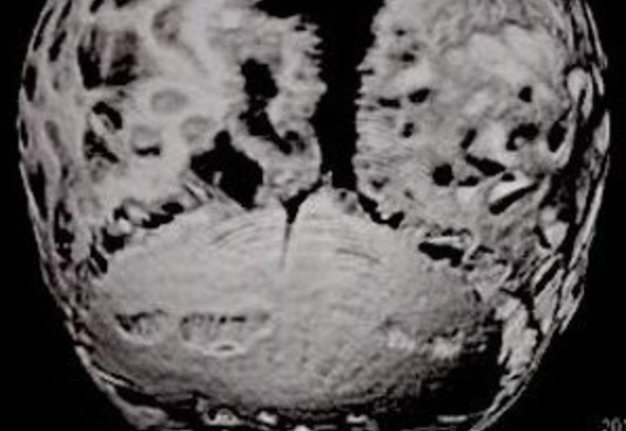
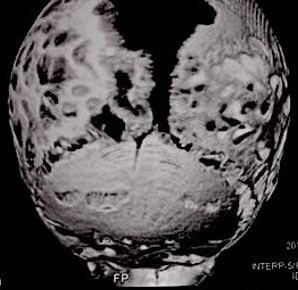
FIGURE 2B. 3D reconstruction CT scans of the skull: posterior view (B). The increase of the space of metopic suture (asterisk) is noted.

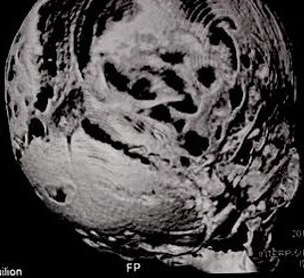
FIGURE 2C. 3D reconstruction CT scans of the skull: left posterolateral view (C). The increase of the space of metopic suture (asterisk) is noted.
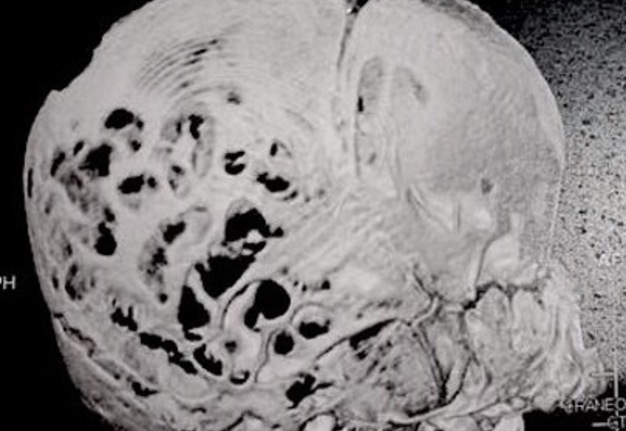
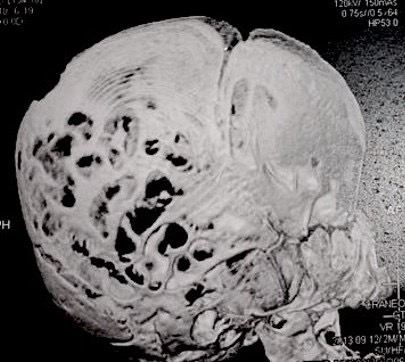
FIGURE 2D. 3D reconstruction CT scans of the skull: left lateral view (D).
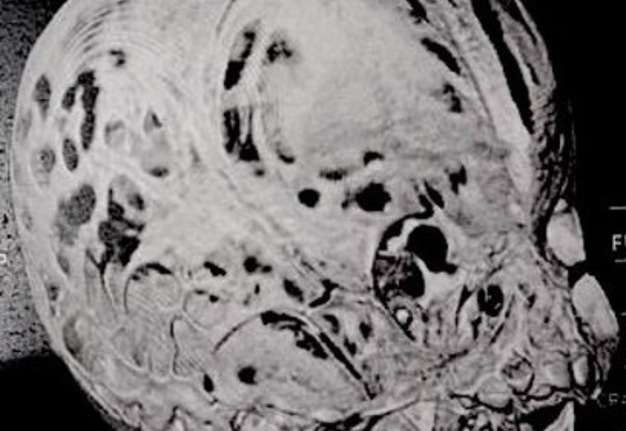
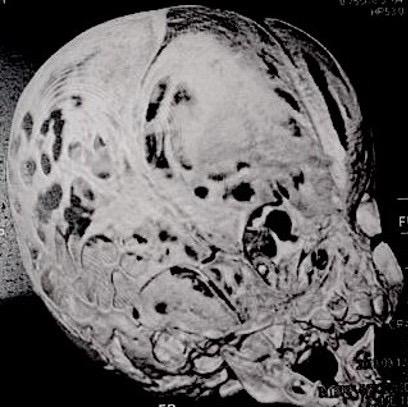
FIGURE 2E. 3D reconstruction CT scans of the skull: left anterolateral view (E).
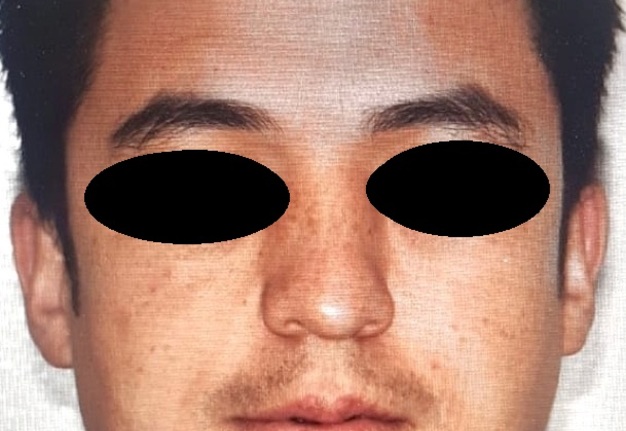
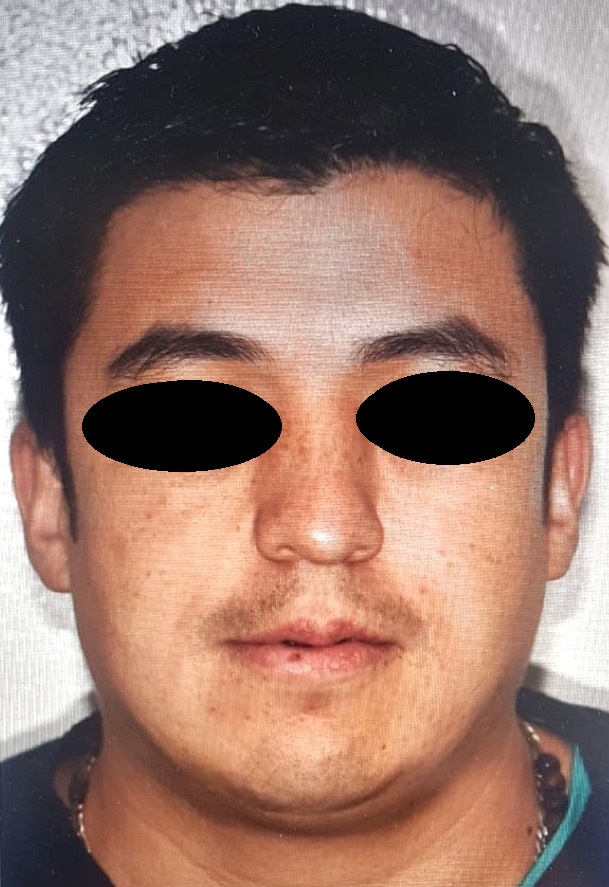
FIGURE 3. Patient´s father. Frontal view with broad forehead, brachycephaly, facial asymmetry (hypertelorism, oblique palpebral fissures directed downward, scarce eyebrows, palpebral ptosis), flat philtrum, thin upper lip, and micrognathia. Slight expression of MS related to the son.
People with Muenke syndrome have the lowest incidence of cleft palate among the most common craniosynostosis syndromes. However, the high and/or cleft palate has been reported in some cohorts,4, 17 therefore, orofacial anomalies should merit clinical attention, since palatal alterations cause interference with the tensor veli palatini muscle and predispose to recurrent and long-term chronic otitis media with increased risk of conductive hearing loss, among other complications.
The cranial surgical procedure is often delayed until facial growth is completed, decreasing the rate of reoperation by excessive frontal bulging. Affected individuals with significant degrees of orbital hypertelorism will require medial accommodation of their orbits through surgical osteotomies, a procedure carried out beyond 5 years of age to ensure adequate surgical correction. Malocclusion`s problems are treated with maxillary and orthodontic orthopedics to allow establish an adequate occlusion. As the occlusion is related to the position of the jaw and maxilla, a treatment plan must be formulated not only to achieve a normal dental occlusion, but also to optimize the aesthetic result of the individual's appearance.
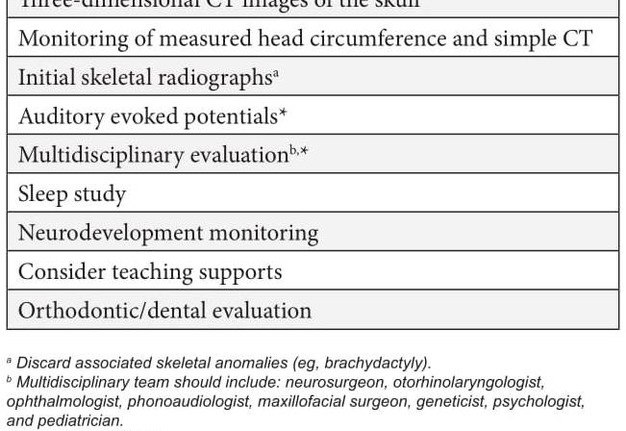
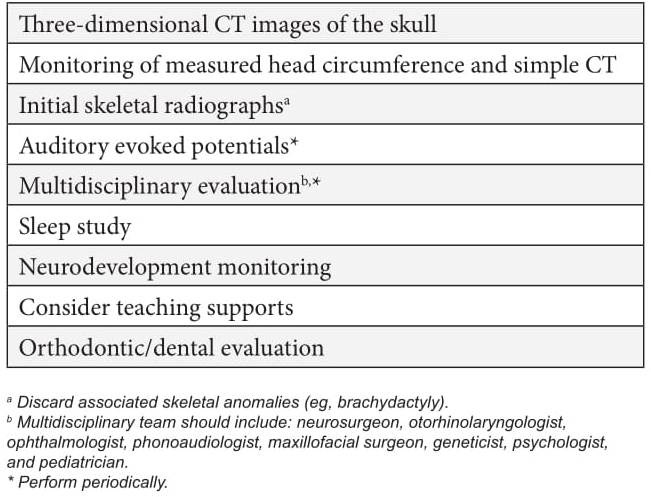
TABLE 1. Clinical Management Suggested in Individuals With Muenke Syndrome.
Historically, the care of individuals with syndromic craniosynostosis has focused purely on the correction of the cranial defect and the monitoring of neurological, ocular and auditory complications. However, the literature and this case demonstrate the importance of other associated anomalies, which is why it is suggested in all cases of craniosynostosis to perform routine oral examination and orthodontic evaluations, as well as to follow the clinical management suggested in Table 1.
Given the variable expressivity of the MS and the reduced penetrance of the gene, it is recommended that the parents of the patients be carefully evaluated by radiographic and molecular study to identify the affected relatives. The genetic risk of recurrence depends on the state of the parents with respect to the variant, if a parent has the pathogenic variant FGFR3 p.Pro250Arg, the risk of inheriting this variant to their offspring is 50%. When the parents are not affected clinically and do not have the pathogenic variant, the risk for the offspring is low, but not null, due to the possibility of germinal mosaicism. The proband's offspring have 50% risk of inheriting the variant.
ROLE OF THE CO-AUTHORS
The co-authors are eaqully contributed to that paper. All of them read and approved the final manuscript.
FUNDINGS
No funding was received for this study.
ACKNOWLEDGEMENTS
We thank to the patient and his family for their kind cooperation and their permission to publish this report.
REFERENCES (39)
-
Muenke M, Gripp K, McDonald-McGinn D, Gaudenz K, Whitaker LA, Bartlett SP, Markowitz RI, Robin NH, Nwokoro N, Mulvihill JJ, Losken HW, Mulliken JB, Guttmacher AE, Wilroy RS, Clarke LA, Hollway G, Adès LC, Haan EA, Mulley JC, Cohen MM Jr, Bellus GA, Francomano CA, Moloney DM, Wall SA, Wilkie AO. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 1997;60(3):555–64.
-
Kruszka P, Addissie Y, Yarnell C, Hadley D, Guillen Sacoto M, Platte P, Paelecke Y, Collmann H, Snow N, Schweitzer T, Boyadjiev SA, Aravidis C, Hall SE, Mulliken JB, Roscioli T, Muenke M. Muenke syndrome: an international multicenter natural history study. Am J Med Genet A 2016;170A(4):918–29. Crossref
-
Doherty E, Lacbawan F, Hadley D, Brewer C, Zalewski C, Kim H, Solomon B, Rosenbaum K, Domingo DL, Hart TC, Brooks BP, Immken L, Lowry RB, Kimonis V, Shanske AL, Jehee FS, Bueno MR, Knightly C, McDonald-McGinn D, Zackai EH, Muenke M. Muenke syndrome (FGFR3-related craniosynostosis): expansion of the phenotype and review of the literature. Am J Med Genet A 2007;143A(24):3204–15. Crossref
-
Agochukwu N, Solomon B, Doherty E, Muenke M. The palatal and oral manifestations of Muenke syndrome. (FGFR3 related craniosynostosis). J Craniofac Surg 2012;23(3):664–8. Crossref
-
Buchanan E, Xue A, Hollier L. Craniofacial syndromes. Plast Reconstruct Surg 2014; 134(1):128–53. Crossref
-
Brookes C, Golden B, Turvey T. Craniosynostosis syndromes. Atlas Oral Maxillofacial Surg Clin N Am 2014;22(2):103–10. Crossref
-
Golla A, Lichmer P, von Gernet S, Winterpacht A, Fairley J, Murken J, Schuffenhauer S. Phenotypic expression of the fibroblast growth factor receptor 3 (FGFR3) mutation P250R in a large craniosynostosis family. J Med Genet 1997;34(8):683–4.
-
Gripp K, McDonald-McGinn D, Gaudenz K, Whitaker L, Bartlett SP, Glat PM, Cassileth LB, Mayro R, Zackai EH, Muenke M. Identification of a genetic cause for isolated unilateral coronal synostosis: a unique mutation in the fibroblast growth factor receptor 3. J Pediatr 1998;132(4):714–6.
-
Sabatino G, Di Rocco F, Zampino G, Tamburrini G, Caldarelli M, Di Rocco C. Muenke syndrome. Childs Nerv Syst 2004;20(5):297–301.
-
Robin NH, Scott JA, Cohen AR, Goldstein JA. Nonpenetrance in FGFR3-associated coronal synostosis syndrome. Am J Med Genet 1998;80(3):296–7.
-
Moloney D, Wall S, Ashworth G, Oldridge M, Glass I, Francomano C, Muenke M, Wilkie AO. Prevalence of Pro250Arg mutation of fibroblast growth factor receptor 3 in coronal craniosynostosis.. Lancet 1997;349(9058):1059–62.
-
Thomas GP, Wilkie AO, Richards PG, Wall SA. FGFR3 P250R mutation increases the risk of reoperation in apparent ‘nonsyndromic’ coronal craniosynostosis. J Craniofac Surg 2005;16(3):347–54. Crossref
-
Rannan-Eliya S, Taylor I, de Heer M, van den Ouweland A, Wall S, Wilkie A. Paternal origin of FGFR3 mutations in Muenke-type craniosynostosis. Hum Genet 2004;115:200–7. Crossref
-
Yoo H, Rah D, Kim Y. Outcome analysis of cranial molding therapy in nonsynostotic plagiocephaly. Archives of Plastic Surgery 2012;39(4):338–44. Crossref
-
Posnick J, Ruiz R, Tiwana P. Craniofacial dysostosis syndromes: stages of reconstruction. Oral Maxillofacial Surg Clin N Am 2004;16(4):475–91. Crossref
-
Escobar L, Hiett A, Marnocha A. Significant phenotypic variability of Muenke syndrome in identical twins. Am J Med Genet A 2009; 149A(6):1273–6. Crossref
-
Reardon W, Wilkes D, Rutland P. Craniosynostosis associated with FGFR3 pro250arg mutation results in a range of clinical presentations including unisutural sporadic craniosynostosis. J Med Genet 1997;34:632–6.
-
Liang W, Nishino I. Lipid storage myopathy. Curr Neurol Neurosci Rep 2011;11:97–103. Crossref
-
Zimmermann R, Strauss J, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004;306(5700):1383–6. Crossref
-
Fischer J, Lefevre C, Morava E, Mussini JM, Laforêt P, Negre-Salvayre A, Lathrop M, Salvayre R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet 2007;39(1):28–30. Crossref
-
Tavian D, Missaglia S, DiMauro S, Bruno C, Pegoraro E, Cenacchi G, Coviello D, Angelini C. A late-onset case of neutral lipid storage disease with myopathy, dropped head syndrome, and peripheral nerve involvement. J Genet Syndr Gene Ther 2013;4(10):e198. Crossref
-
Jocken J, Smit E, Goossens G, Essers YP, van Baak MA, Mensink M, Saris WH, Blaak EE. Adipose triglyceride lipase (ATGL) expression in human skeletal muscle is type I (oxidative) fiber specific. Histochem Cell Biol 2008;129:535–8. Crossref
-
Chanarin I, Patel A, Slavin G, Wills E, Andrews T, Stewart G. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J 1975;5957(1):553–5.
-
Reilich P, Horvath R, Krause S, Schramm N, Turnbull DM, Trenell M, Hollingsworth KG, Gorman GS, Hans VH, Reimann J, MacMillan A, Turner L, Schollen A, Witte G, Czermin B, Holinski-Feder E, Walter MC, Schoser B, Lochmüller H. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J Neurol 2011;258(11):1987–97. Crossref
-
Soni K, Mardones G, Sougrat R, Smirnova E, Jackson C, Bonifacino J. Coatomer-dependent protein delivery to lipid droplets. J Cell Sci 2009;122(11):1834–41. Crossref
-
Ash D, Papadimitriou D, Hays A, DiMauro S, Hirano M. A novel mutation in PNPLA2 leading to neutral lipid storage disease with myopathy. Arch Neurol 2012;69(9):1190–2. Crossref
-
Ohkuma A, Nonaka I, Malicdan M, Noguchi S, Ohji S, Nomura K, Sugie H, Hayashi YK, Nishino I. Distal lipid storage myopathy due to PNPLA2 mutation. Neuromuscul Disord 2008;18:671–4. Crossref
-
Akiyama M, Sakai K, Ogawa M, McMillan J, Sawamura D, Shimizu H. Novel duplication mutation in the patatin domain of adipose triglyceride lipase (PNPLA2) in neutral lipid storage disease with severe myopathy. Muscle Nerve 2007;36:856–9. Crossref
-
Campagna F, Nanni L, Quagliarini F, Pennisi E, Michailidis C, Pierelli F, Bruno C, Casali C, DiMauro S, Arca M. Novel mutations in the adipose triglyceride lipase gene causing neutral lipid storage disease with myopathy. Biochem Biophys Res Commun 2008;377(3):843–6. Crossref
-
Kobayashi K, Inoguchi T, Maeda Y, Nakashima N, Kuwano A, Eto E, Ueno N, Sasaki S, Sawada F, Fujii M, Matoba Y, Sumiyoshi S, Kawate H, Takayanagi R. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J Clin Endocrinol Metab 2008;93(7):2877–84. Crossref
-
Akman H, Davidzon G, Tanji K, Macdermott E, Larsen L, Davidson MM, Haller RG, Szczepaniak LS, Lehman TJ, Hirano M, DiMauro S. Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromuscul Disord 2010;20(6):397–402. Crossref
-
Fiorillo C, Brisca G, Cassandrini D, Scapolan S, Astrea G, Valle M, Scuderi F, Trucco F, Natali A, Magnano G, Gazzerro E, Minetti C, Arca M, Santorelli FM, Bruno C. Subclinical myopathy in a child with neutral lipid storage disease and mutations in the PNPLA2 gene. Biochem Biophys Res Commun 2013;430(1):241–4. Crossref
-
Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006;312(5774):734–7. Crossref
-
Cree M, Newcomer B, Read L, Shef eld-Moore M, Paddon- Jones D, Chinkes D, Aarsland A, Wolfe RR. Plasma triglycerides are not related to tissue lipids and insulin sensitivity in elderly following PPAR-alpha agonist treatment. Mech Ageing Dev 2007;128(10):558–65. Crossref
-
van de Weijer T, Havekes B, Bilet L, Hoeks J, Sparks L, Bosma M, Paglialunga S, Jorgensen J, Janssen MC, Schaart G, Sauerwein H, Smeets JL, Wildberger J, Zechner R, Schrauwen-Hinderling VB, Hesselink MK, Schrauwen P. Effects of bezafibrate treatment in a patient and a carrier with mutations in the PNPLA2 gene, causing neutral lipid storage disease with myopathy. Circ Res 2013;112(5):e51–e54. Crossref
-
Laforêt P, Stojkovic T, Bassez G, Carlier P, Clément K, Wahbi K, Petit FM, Eymard B, Carlier RY. Neutral lipid storage disease with myopathy: a whole-body nuclear MRI and metabolic study. Mol Genet Metab 2013;108:125–31. Crossref
-
Laforet P, Vianey-Saban C. Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscul Disord 2010;20(11):693–700. Crossref
-
Bruno C, Dimauro S. Lipid storage myopathies. Curr Opin Neurol 2008;21(5):601–6. Crossref
-
Janssen M, van Engelen B, Kapusta L, Lammens M, van Dijk M, Fischer J, van der Graaf M, Wevers RA, Fahrleitner M, Zimmermann R, Morava E. Symptomatic lipid storage in carriers for the PNPLA2 gene. Eur J Hum Genet 2013;21(8):807–15. Crossref